Theory and Simulation
Members use first-principles theoretical and computational methods to investigate materials with applications in next-generation optoelectronics and quantum technologies. Methods include density functional theory (DFT) and time-dependent DFT, machine-learning accelerated molecular dynamics and structure predictions, and many-body perturbation theory approaches. Often in collaboration with experimental researchers, we focus on excited-state properties and finite-temperature effects, including the influence of lattice vibrations (phonons) and polarons, defects, anharmonicity, and disorder, as well as structural phase transitions, charge and thermal transport.
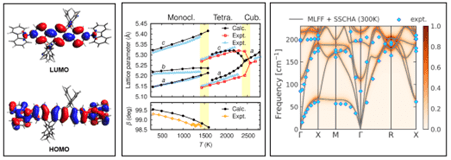
Figure 1 (a) End-capped oligophenylene molecule as a deep blue lasing material investigated by TDDFT. (b) Accurate structural phase transitions from machine-learned force fields and molecular dynamics simulations. (c) Temperature-dependent anharmonic phonons in a perovskite material.
References:
- ACS Applied Materials & Interfaces 2024, 16, 46506.
- npj Computational Materials 2021, 7, 156.
- Physical Review Letters 2019, 122, 246403.
- Physical Review Materials 2024, 8, 104603.